




高风险性:许多生物材料无法通过最终灭菌(如湿热、辐射)处理。
过程开放性:细胞培养、纯化、灌装等环节存在开放性操作,污染风险高。
污染不可逆:培养周期长,一旦污染,整批次失败。
因此,疫苗生产必须遵循2010年修订版GMP(与欧盟GMP高度接轨),并接受《中国药典》在无菌、微生物限度等方面的补充约束,形成双重标准监管体系。

根据GMP,洁净区分为A、B、C、D四级。疫苗生产关键过程(配制、过滤、灌装)通常在B级背景下的A级操作区进行。
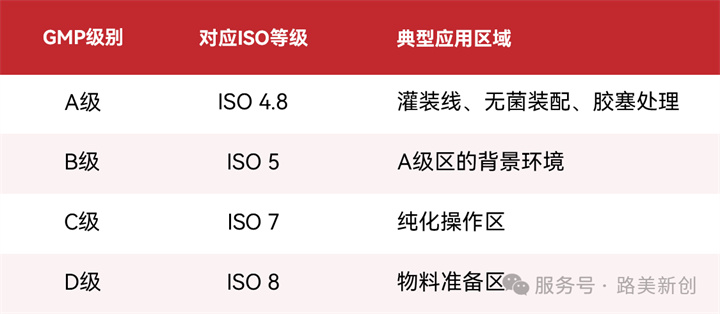
-GMP:规定洁净区分级、动态/静态监测、微生物限度及确认周期(如A/B级区再确认最长6个月,C/D级12个月)。
《中国药典》:详述无菌检查、微生物限度、内毒素等具体方法。
- 2022年版欧盟GMP附录1《无菌药品生产》的理念(基于风险、历史数据)已深度影响我国要求,强调建立全面的环境监测计划。

A级要求极其严苛:静态和动态下,≥0.5μm粒子≤3520个/m³,而≥5.0μm粒子仅允许20个/m³。各级别具体限值如下:
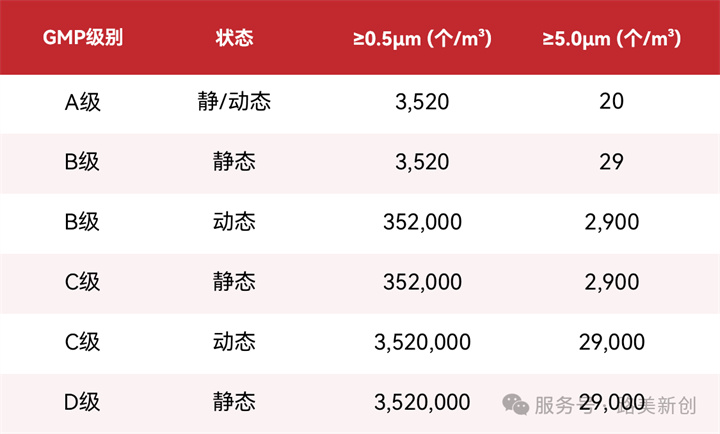
注:我国GMP对A级≥5.0μm限值(20个/m³)严于欧盟GMP附录1(29个/m³)。

- A级关键区必须实现全过程动态监测,尤其是灌装线。
- 日常监测需满足**数据完整性**(审计追踪、权限管理、电子签名),符合21 CFR Part 11等规范。
- 仪器需具备抗VHP(汽化过氧化氢)消毒能力,适应洁净室熏蒸环境。
- 采样流量通常为28.3L/min,以便快速完成A级区1000L采样。

疫苗生产对微生物控制极严,A级区要求“无生长”(我国GMP规定<1 CFU)。各级别限度如下:
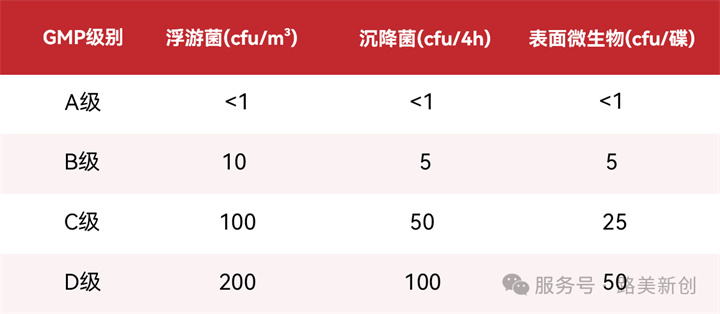
-采样方法:按GB/T 16293,宜采用撞击法机理的采样器(如狭缝式、离心式),确保捕获效率。
采样量:A/B级单次采样不少于1000L。
测试状态:分别进行静态和动态测试。
沉降菌:暴露时间应基于回收率研究确定。
-仪器应便于远程控制,减少人为干扰,且耐受常规消毒剂。

A级单向流洁净区断面风速需控制在**0.36-0.54 m/s。此范围既能有效带走污染粒子,又不干扰操作。
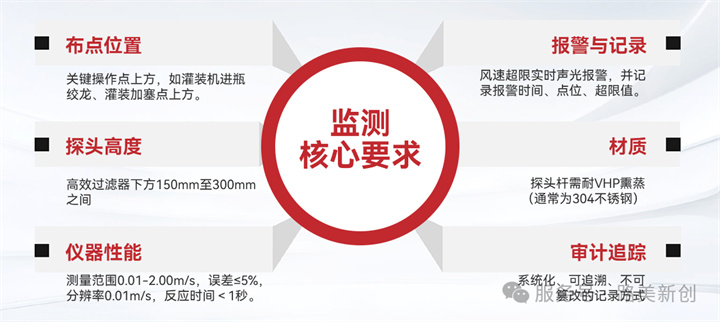
5.2 监测核心要求
布点位置:关键操作点上方,如灌装机进瓶绞龙、灌装加塞点上方。
探头高度:高效过滤器下方150mm至300mm之间。
仪器性能:测量范围0.01-2.00m/s,误差≤5%,分辨率0.01m/s,反应时间<1秒。
报警与记录:风速超限实时声光报警,并记录报警时间、点位、超限值。
材质:探头杆需耐VHP熏蒸(通常为304不锈钢)。

- 不同洁净级别之间、洁净区与非洁净区之间压差应≥10 Pa。
- A/B级区压差需实时监测并设报警;可经验证设置报警延时,避免正常开关门误报。

- 温度:18℃~26℃;相对湿度:45%~65%。
- 控制目标兼顾抑制微生物生长和人员舒适性。

- 基于工艺关键性、设备、操作步骤及历史数据,开展风险分析。
- 确定取样点数量/位置、取样频率(关键区域应高频动态监测)。
- 欧盟GMP附录1明确要求:A/B级再确认最长6个月,C/D级最长12个月。
7.2 异常处理基本流程
当监测数据接近或超过限度时:
1. 立即通知责任人。
2. 调查根本原因(如人员操作、过滤器泄漏、消毒失效)。
3. 评估对已生产产品的影响。
4. 制定纠正与预防措施(CAPA)。
5. 完整记录全过程,确保可追溯。

(注:本文聚焦科普与标准解析,具体设备选型请根据实际工艺需求和合规要求评估。)



